

Abstract
Introduction:
Antiphospholipid syndrome (APS) is an autoimmune disorder caused by "antiphospholipid" antibodies (aPL) directed against β2-glycoprotein I (β2GPI). In the presence of β2GPI, aPL activate endothelial cells through TLR4-mediated and other pathways. Previous studies suggest that a region encompassing amino acids 39-43 of β2GPI domain 1 comprises an important binding site for pathogenic aPL. However, due to difficulty in expressing full-length recombinant β2GPI, antibody specificity has only been tested using individual, recombinant β2GPI domains. Further definition of antibody specificity using intact recombinant β2GPI provides a new approach for diagnostic and mechanistic studies. In the present study we describe the expression of full-length, wild-type recombinant β2GPI and site-directed mutants encompassing the putative β2GPI binding region in domain 1. Binding of APS patient-derived anti-β2GPI antibodies to these proteins and their ability to support anti-β2GPI-mediated endothelial cell activation was determined.
Methods:
Full length APOH cDNA with a modified signal peptide (spm) was cloned into the lentiviral vector pLenti CMV Puro DEST. We generated two mutants spanning the 39 to 43 amino acid region: SEGVG (R39S; G40E; M42V; R43G) and AAGMA (R39A; G40A; R43A). The final recombinant plasmids were termed as pDEST-spmAPOH-WT; pDEST-spmAPOH-SEGVG and pDEST-spmAPOH-AAGMA. Lentivirus was produced using the Lentiviral Gateway Expression kit using GP2-293 (HEK) cells. Stable cell lines were generated by transducing HEK cells with APOH-lentivirus and selecting against puromycin. Stable cell lines were transferred to serum-free media and grown in suspension. Cell culture supernatants containing secreted β2GPI were filtered and concentrated, and recombinant β2GPI was purified using a hitrap-heparin column.
Anti-β2GPI antibody binding to plasma-derived and rβ2GPI was determined using a standard β2GPI ELISA and SPR (Surface Plasmon Resonance) on a Biacore 3000. Biosensor analysis was performed by crosslinking β2GPI to carboxymethyl-dextran coated sensor chips using amine coupling. Anti-β2GPI antibodies at concentrations ranging from 1-15,000 nM, were flowed through channels until equilibrium binding was achieved, at which point dissociation was assessed over a 10 minute interval. The BIAevaluation program was used to calculate association and dissociation rates. Endothelial cell activation was assessed by measuring cell surface E-selectin expression after incubating cells with control IgG or anti-β2GPI antibodies in the presence or absence of wild-type or mutant rβ2GPI. Molecular modeling was performed using PyMol.
Results:
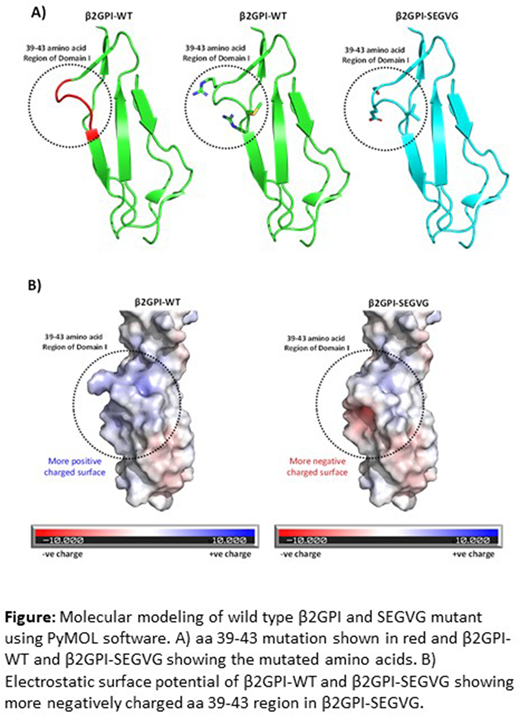
After transduction with lentivirus encoding β2GPI with a modified signal peptide, HEK-293 cells were able to express and efficiently secrete rβ2GPI and site-directed mutants. Using a standard β2GPI-ELISA and SPR we found no significant differences in binding affinity of aPL towards plasma-derived and wild-type rβ2GPI. However, in the anti-β2GPI ELISA, binding of patient-derived aPL (APS21) to both mutants was reduced by ~70% compared to wild-type rβ2GPI. Consistent with binding specificity for a region encompassing aa 39-43, the Kd for binding of APS21 anti-β2GPI antibodies to wild-type and rβ2GPI-SEGVG determined by SPR was 20 nM and 5000 nM, respectively. Moreover, the ability of patient-derived APS21 anti-β2GPI antibodies to activate endothelial cells was reduced by >60% in the presence of rβ2GPI-SEGVG compared to wild-type rβ2GPI. Molecular modeling of β2GPI demonstrated that a mutation in the aa 39-43 region is predicted to cause a change from a net positive to a net negative charge without any structural change (Figure 1).
Conclusion:
These studies are the first to assess binding of human anti-β2GPI antibodies to rβ2GPI and site-directed mutants expressed in mammalian cells. No significant differences between binding of aPL to plasma derived and wild-type rβ2GPI were observed. APS21 aPL has specificity towards epitope 39-43 of β2GPI domain 1 and its binding affinity to rβ2GPI-SEGVG was significantly reduced. Functional studies demonstrate the importance of β2GPI aa 39 to 43 in supporting endothelial cell activation by anti-β2GPI antibodies. These recombinant proteins should facilitate further studies concerning the role of aPL-β2GPI interactions in the diagnosis and pathogenesis of APS.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal